擴(kuò)增子測(cè)序是對(duì)特定長(zhǎng)度的PCR產(chǎn)物或者捕獲的片段進(jìn)行測(cè)序。16S/18S/ITS等擴(kuò)增子測(cè)序即通過(guò)提取環(huán)境樣品的DNA,選擇合適的通用引物擴(kuò)增16S/18S/ITS的某一或某幾個(gè)區(qū),使用Illumina MiSeq/HiSeq將目的區(qū)域正反向讀通,通過(guò)檢測(cè)目的區(qū)域的序列變異和豐度,對(duì)環(huán)境樣本物種分類及豐度、種群結(jié)構(gòu)、系統(tǒng)進(jìn)化、群落比較等方面信息進(jìn)行分析的研究方法。
宏基因組 ( Metagenome)(也稱微生物環(huán)境基因組Microbial Environmental Genome, 或元基因組)是由 Handelsman 等 1998 年提出的新名詞, 其定義為“the genomes of the total microbiota found in nature” , 即環(huán)境中全部微小生物遺傳物質(zhì)的總和。它包含了可培養(yǎng)的和未可培養(yǎng)的微生物的基因,目前主要指環(huán)境樣品中的細(xì)菌和真菌的基因組總和。宏基因組學(xué)(或元基因組學(xué),metagenomics)是一種以環(huán)境樣品中的微生物群體基因組為研究對(duì)象,以功能基因篩選和/或測(cè)序分析為研究手段,以微生物多樣性、種群結(jié)構(gòu)、進(jìn)化關(guān)系、功能活性、相互協(xié)作關(guān)系及與環(huán)境之間的關(guān)系為研究目的的新的微生物研究方法。一般包括從環(huán)境樣品中提取基因組DNA, 進(jìn)行高通量測(cè)序分析,或克隆DNA到合適的載體,導(dǎo)入宿主菌體,篩選目的轉(zhuǎn)化子等工作。
特定生物種基因組研究使人們的認(rèn)識(shí)單元實(shí)現(xiàn)了從單一基因到基因集合的轉(zhuǎn)變,宏基因組研究將使人們擺脫物種界限,揭示更高更復(fù)雜層次上的生命運(yùn)動(dòng)規(guī)律。在目前的基因結(jié)構(gòu)功能認(rèn)識(shí)和基因操作技術(shù)背景下,細(xì)菌宏基因組成為研究和開發(fā)的主要對(duì)象。細(xì)菌宏基因組、細(xì)菌人工染色體文庫(kù)篩選和基因系統(tǒng)學(xué)分析使研究者能更有效地開發(fā)細(xì)菌基因資源,更深入地洞察細(xì)菌多樣性。
全基因組測(cè)序分為從頭測(cè)序(de novo sequencing)和重測(cè)序(re-sequencing)。
從頭測(cè)序(de novo)不需要任何參考基因組信息即可對(duì)某個(gè)物種的基因組進(jìn)行測(cè)序,利用生物信息學(xué)分析方法進(jìn)行拼接、組裝,獲得該物種的基因組序列圖譜,從而推進(jìn)該物種的后續(xù)研究。
基因組重測(cè)序是對(duì)有參考基因組物種的不同個(gè)體進(jìn)行的基因組測(cè)序,并在此基礎(chǔ)上對(duì)個(gè)體或群體進(jìn)行差異性分析。基因組重測(cè)序主要用于輔助研究者發(fā)現(xiàn)單核苷酸多態(tài)性位點(diǎn)(SNPs)、拷貝數(shù)變異(CNV)、插入/缺失(Indel)等變異類型,以較低的價(jià)格將單個(gè)參考基因組信息擴(kuò)增為生物群體的遺傳特征。全基因組重測(cè)序在人類疾病和動(dòng)植物育種研究中廣泛應(yīng)用。
微生物是地球上最豐富、最多樣的生命形式,它們是所有生態(tài)系統(tǒng)的重要組成部分,在醫(yī)療健康領(lǐng)域和工業(yè)應(yīng)用領(lǐng)域中發(fā)揮著至關(guān)重要的作用。微生物基因組學(xué)研究在醫(yī)學(xué)上可應(yīng)用于致病相關(guān)基因的鑒定、開發(fā)新型抗生素等,而在生物技術(shù)上可用于生物降解、酶工業(yè)、食品生物以及抗生物質(zhì)的研究,同時(shí)對(duì)進(jìn)化、功能預(yù)測(cè)等方面也有著重要意義。
自1994年美國(guó)發(fā)起微生物基因組研究計(jì)劃(MGP)以來(lái),微生物組學(xué)在生物領(lǐng)域蓬勃發(fā)展,而在2001年中國(guó)繪制出首張微生物基因組“完成圖”以后,隨著高通量測(cè)序技術(shù)的誕生,微生物基因組研究也進(jìn)入了井噴期,我國(guó)先后啟動(dòng)了萬(wàn)種微生物基因組計(jì)劃、百萬(wàn)微生態(tài)系統(tǒng)基因組計(jì)劃等。
研究的深入依賴技術(shù)的進(jìn)步。短讀長(zhǎng)測(cè)序技術(shù)由于無(wú)法跨越富含重復(fù)區(qū)域的基因組使得很多微生物研究止步于基因組草圖,而長(zhǎng)讀長(zhǎng)測(cè)序技術(shù)卻可以輕松獲取細(xì)菌基因組完成圖,同時(shí)在復(fù)雜微生物群落的鑒定中可達(dá)到“種級(jí)別”的鑒定水平。Frank等人于2016年結(jié)合Hiseq2000和PacBio RS II對(duì)沼氣反應(yīng)器內(nèi)微生物宏基因組進(jìn)行研究發(fā)現(xiàn)PacBio技術(shù)能顯著提升組裝水平,而2013年Chin等僅運(yùn)用PacBio就組裝出16個(gè)微生物基因組,與參考基因組一致性達(dá)99.9999%。
如果說(shuō)PacBio技術(shù)為微生物研究打開新世界的大門,那么基于納米孔電流檢測(cè)的ONT技術(shù)則開啟了微生物組學(xué)的黃金時(shí)代。

我們擁有專業(yè)的生信分析團(tuán)隊(duì),除常規(guī)的基礎(chǔ)分析、標(biāo)準(zhǔn)分析外,可根據(jù)您的需求,個(gè)性化定制分析方案、優(yōu)化分析流程,解除您的后顧之憂……
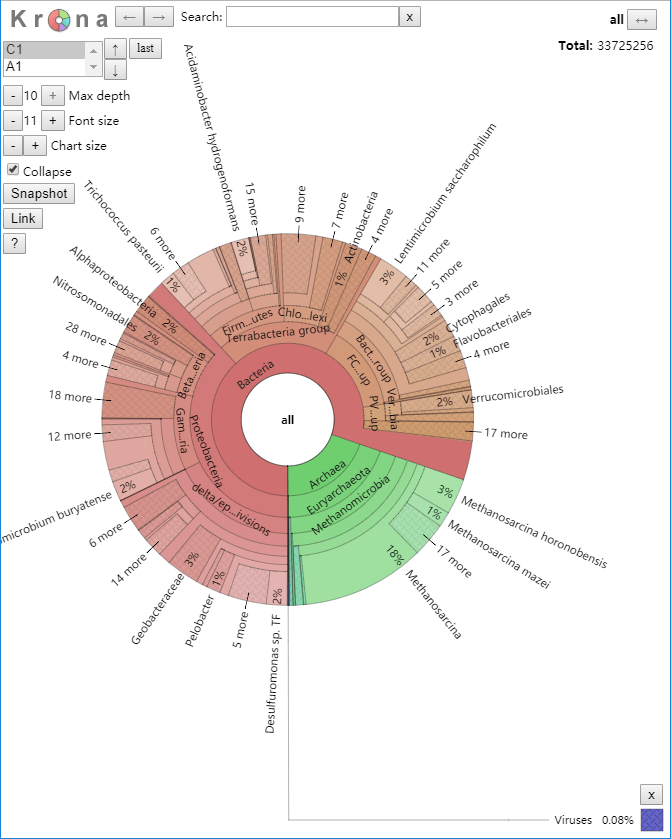
Krona分析,通過(guò)可視化交互形式,直觀展示不同樣本或分組之間的物種多樣性的差異。
線性判別分析(Linear Determinant Analysis, LDA)是對(duì)費(fèi)舍爾的線性鑒別方法的歸納,這種方法使用統(tǒng)計(jì)學(xué)、模式識(shí)別和機(jī)器學(xué)習(xí)方法,試圖找到兩類物體或事件的特征的一個(gè)線性組合,以能夠特征化或區(qū)分它們。所得的組合可用來(lái)作為一個(gè)線性分類器,或者更常見(jiàn)的是,為后續(xù)的分類做降維處理。
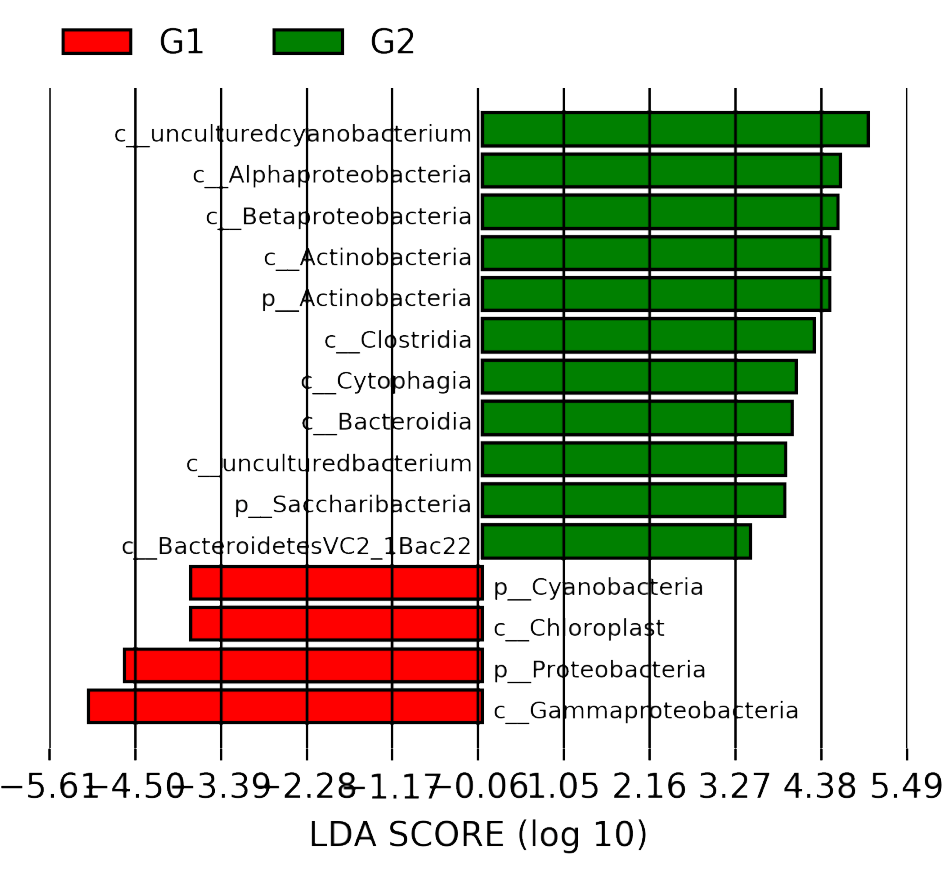
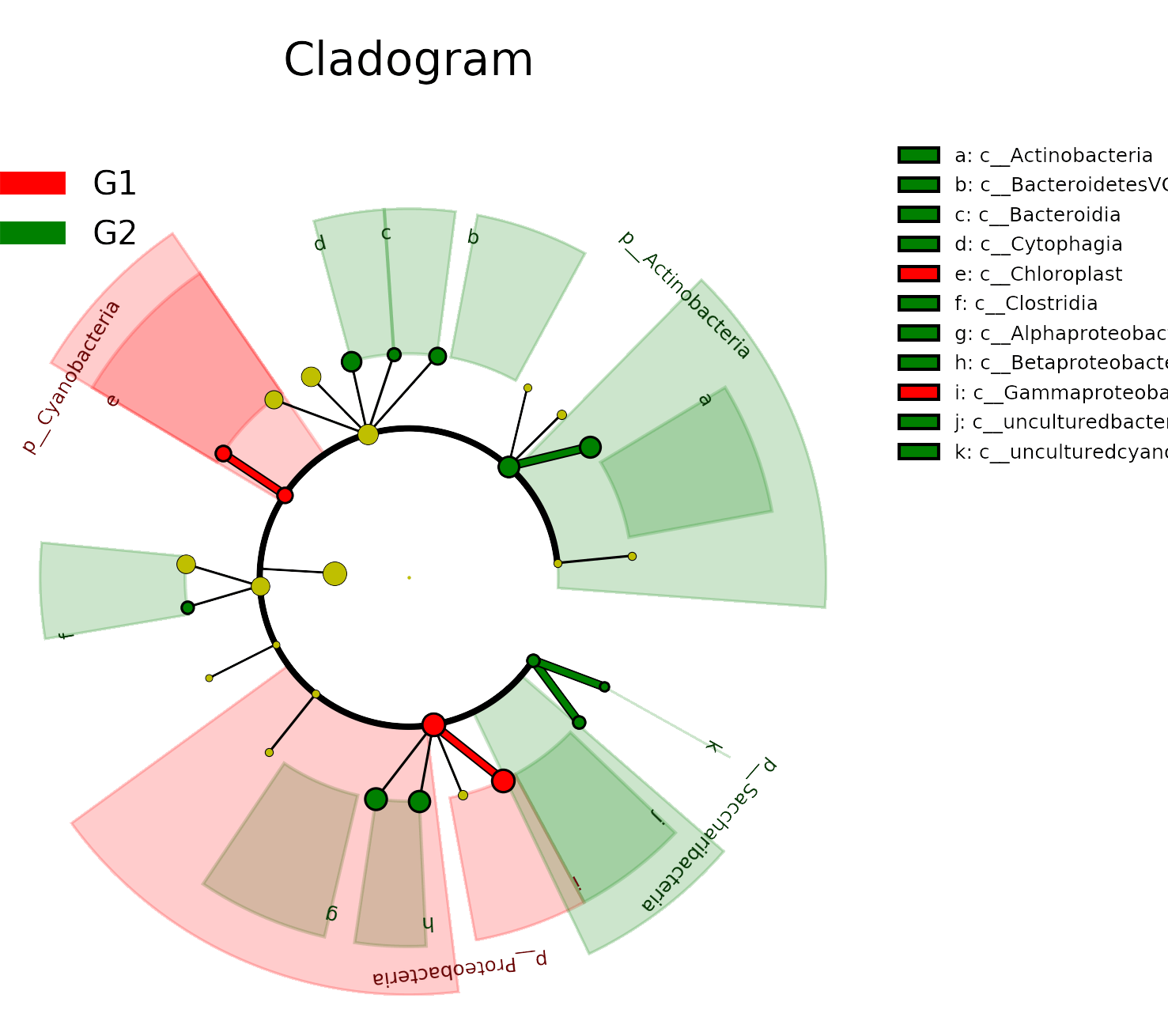
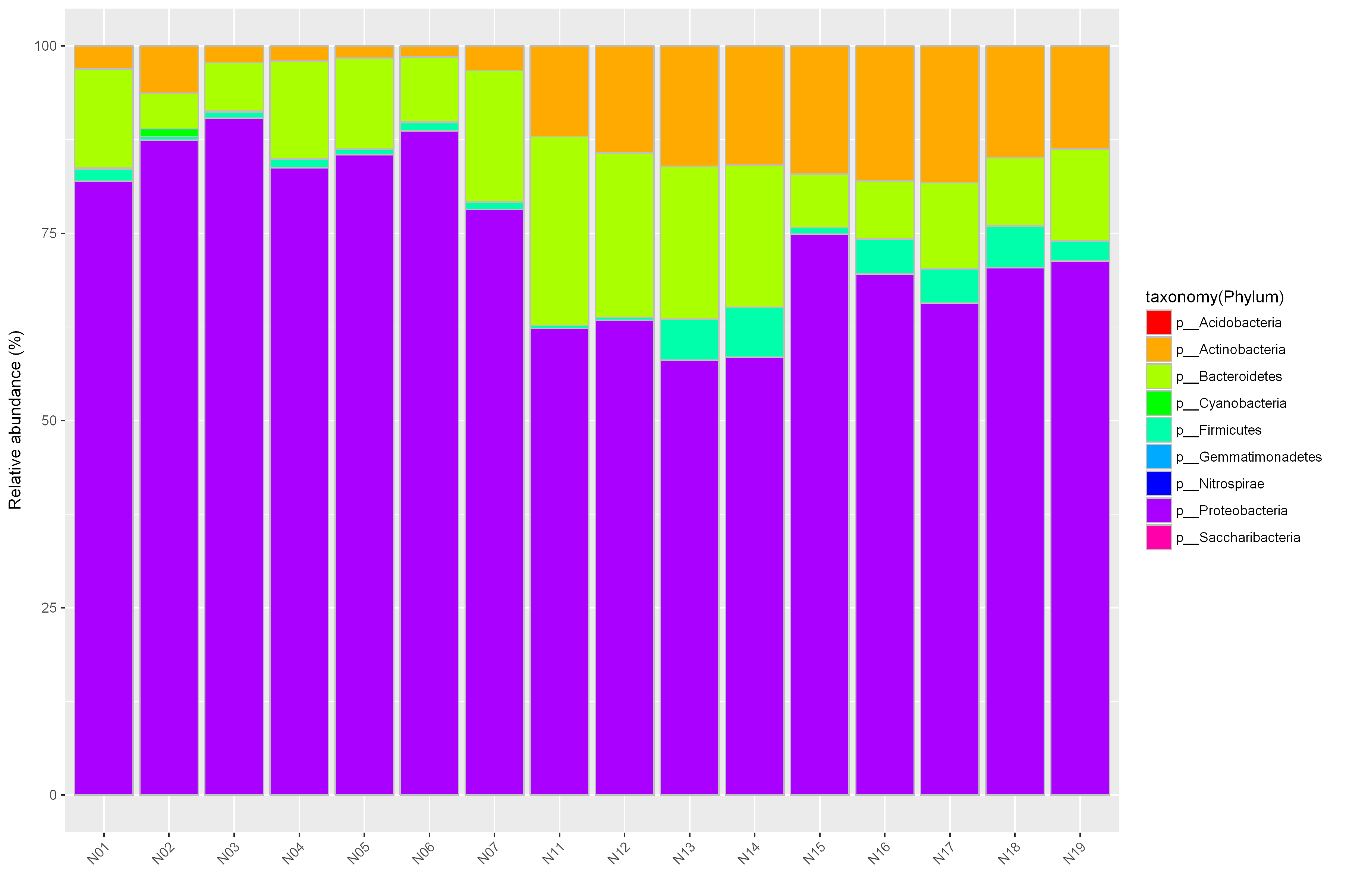
根據(jù)物種的相對(duì)豐度,選取門水平top5、屬水平top15、種水平top20的優(yōu)勢(shì)物種,分析各樣本間優(yōu)勢(shì)物種的分布情況。
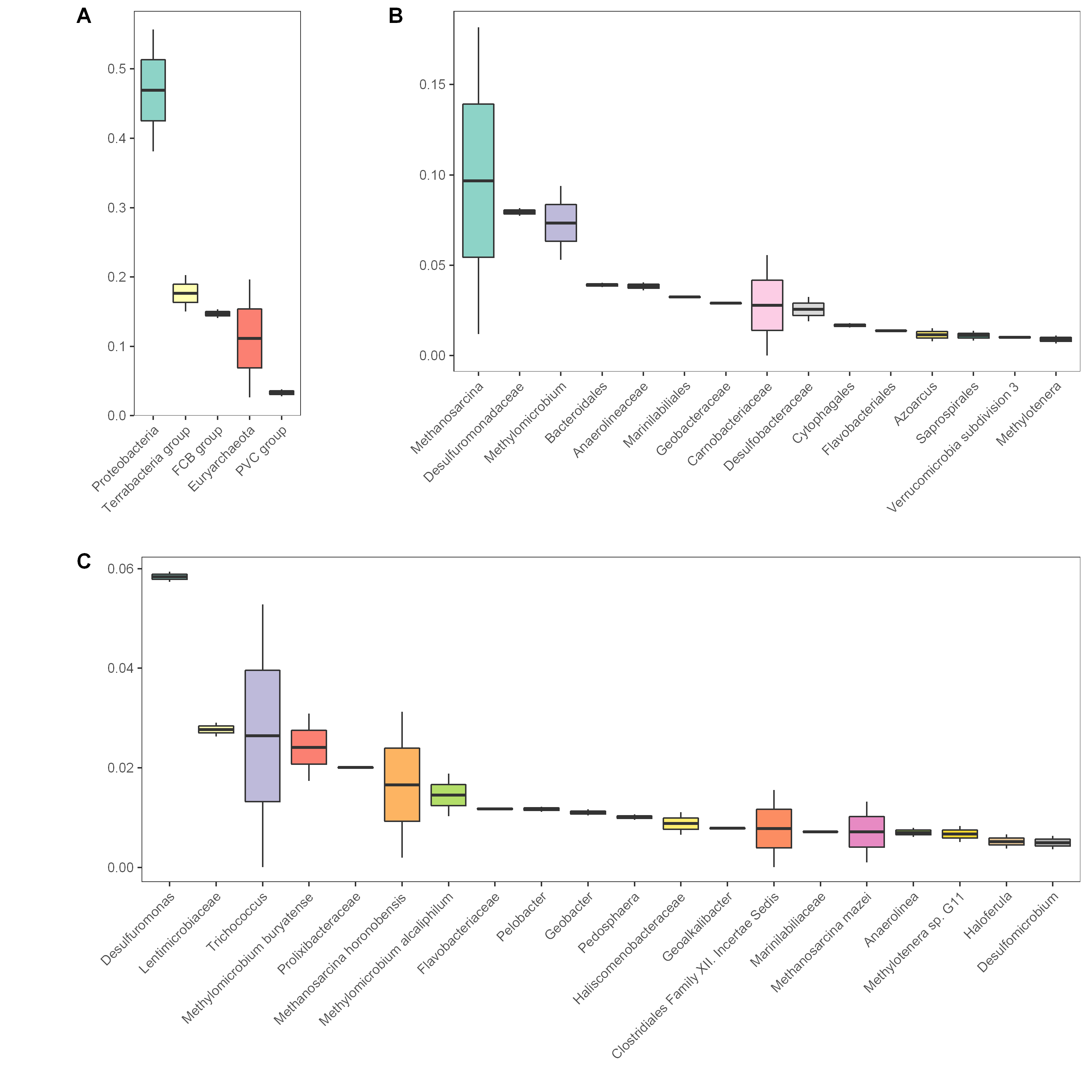
選定一個(gè)或多個(gè)需要分析的樣品,選定一個(gè)分類學(xué)水平,按照相應(yīng)多樣性信息作圖,反映各樣品的群落結(jié)構(gòu)。